
Abstract
Introduction: This article aims to provide a narrative review of the most recent primary literature on the pathways associated with the neuroprotective effects of remote ischaemic post-conditioning (RIPC) in stroke and discuss the prospect for application in the clinical setting.
Summary: This narrative review identified multiple pre-clinical in-vivo studies. These studies found that RIPC modulates a wide variety of pathways within the brain, including those associated with inflammation, reactive oxygen species (ROS) production, antioxidant regulation, and oedema development. Modulation of these pathways was associated with a significant reduction in the neuronal damage, a finding supported by measured reductions in cerebral infarct volume and apoptotic neuronal populations. Clinical research was limited; only one trial on RIPC in stroke was discovered. Whilst the results were positive the small sample size of the study does not make them definitive.
RIPC as an intervention for ischaemia reperfusion injury (IRI) in stroke has been found to be considerably effective in animal models, stimulating a wide variety of neuroprotective pathways. The limited clinical research has not yet been able to confirm RIPC efficacy in human stroke models but should be a catalyst for further research.
Introduction
Cerebrovascular disease constitutes the third greatest cause of mortality in the Australian population, with 23 people dying from a stroke every day [1]. Ischaemic stroke occurs when an artery, supplying a particular area of the brain, is blocked by an embolus, starving neuronal tissue of oxygen and resulting in permanent damage of the local area [2]. Treatment for ischaemic stroke usually involves thrombolysis which dissolves the embolus and restores blood supply to the ischaemic area [1]. Although reperfusion is a critical intervention, it is also associated with a spike in neuronal cell damage, in a phenomena known as ischaemia reperfusion injury (IRI) [2,3]. In recent years, considerable effort has gone into developing ways to limit the impact of IRI. One promising therapy is remote ischaemic conditioning (RIC) [4].
 The principle of RIC is to expose the brain to a series of sub-lethal cycles of ischaemia and reperfusion through intermittent vascular occlusion of another distant and accessible organ such as the upper or lower limb [5]. Transient ischaemia performed in this manner is known to activate several endogenous neuroprotective pathways which reduce the damage associated with an IRI [5,6]. RIC can be performed before the onset of ischaemia (“pre-conditioning”), during the ischaemic event (“per-conditioning”), and after reperfusion (“post-conditioning”) [5]. By their very nature, both pre- and per-conditioning require pre-emptive action which is simply not practical in the health care setting. As such, this review focuses on remote ischaemic post-conditioning (RIPC) performed on the femoral artery in the time immediately following a stroke [7]. This review will explore and integrate the pre-clinical literature surrounding the neuroprotective pathways of RIPC and thus produce a narrative on how this novel intervention acts on a molecular level, and how it may translate to clinical medicine.
The principle of RIC is to expose the brain to a series of sub-lethal cycles of ischaemia and reperfusion through intermittent vascular occlusion of another distant and accessible organ such as the upper or lower limb [5]. Transient ischaemia performed in this manner is known to activate several endogenous neuroprotective pathways which reduce the damage associated with an IRI [5,6]. RIC can be performed before the onset of ischaemia (“pre-conditioning”), during the ischaemic event (“per-conditioning”), and after reperfusion (“post-conditioning”) [5]. By their very nature, both pre- and per-conditioning require pre-emptive action which is simply not practical in the health care setting. As such, this review focuses on remote ischaemic post-conditioning (RIPC) performed on the femoral artery in the time immediately following a stroke [7]. This review will explore and integrate the pre-clinical literature surrounding the neuroprotective pathways of RIPC and thus produce a narrative on how this novel intervention acts on a molecular level, and how it may translate to clinical medicine.
Ischaemia reperfusion injury
IRI is a multi-faceted process, in which reperfusion is associated with cellular death, rather than the restoration of normal function. This abnormal response is a consequence of the ischaemia induced dysregulation of cellular function [8]. One such example is the dysfunction of the Na+/K+ ATPase which triggers the accumulation of intracellular sodium. Sodium accumulation is an initiator for a series of events including calcium overload, excitotoxicity, acidosis, cellular swelling, and the initiation of apoptosis [4].
Another event which occurs during prolonged ischaemia is damage and dysregulation of the mitochondrial electron transport chain (ETC). Once a cell undergoes reperfusion, the ETC will again utilise oxygen to try to produce ATP. However, due to extensive damage, a high proportion of this oxygen will instead be metabolised to form reactive oxygen species (ROS) [9]. The excessive ROS production overwhelms the brain’s anti-oxidant system, resulting in lipid peroxidation, dysregulation of proteins, DNA damage, and alterations in transcription [8].
Ischaemia also has the effect of priming the endothelium for leukocyte recruitment through increasing adhesion molecules and the transcription of pro-inflammatory factors, such as NFκB. Upon reperfusion, these adaptations will stimulate the recruitment of neutrophils to the ischaemic area, where they will release ROS and further exacerbate oxidative damage and cell death [10].

Discussion
HIF1a/TIM3 axis in the initiation of inflammation
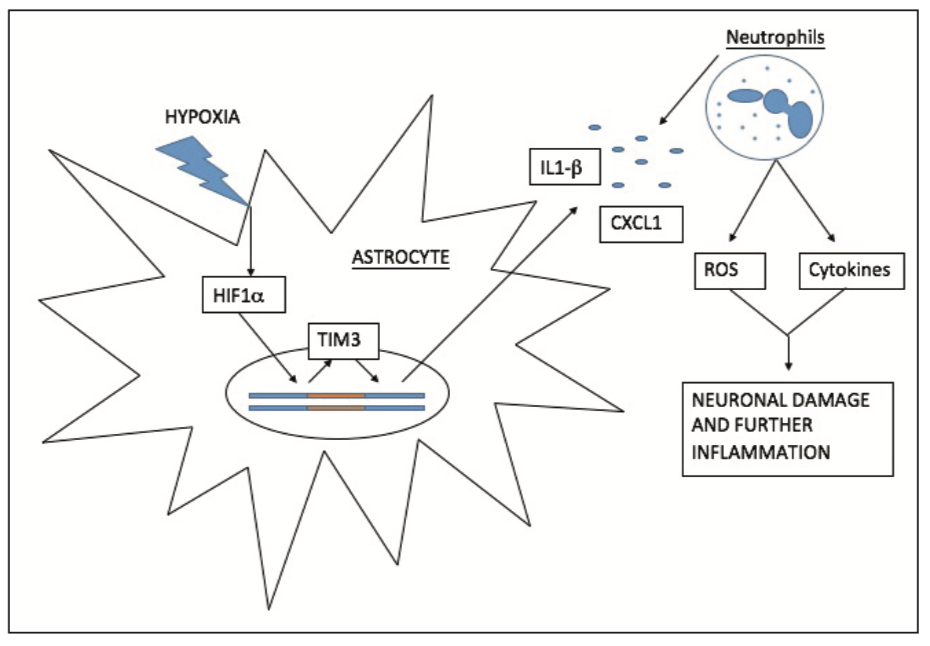
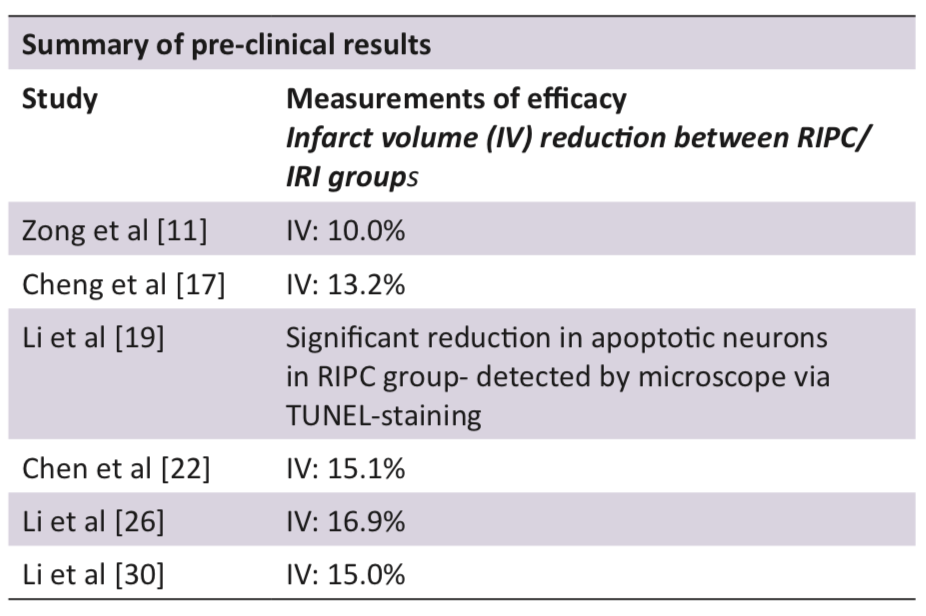
Inflammation is a key contributor to the damage associated with IRI, with the recruitment of leukocytes responsible for the substantial production of ROS and cytokines [10]. The inflammatory response of cerebral IRI is thought to be mediated in part by Hypoxia Inducible Factor 1a (HIF1a), which is activated and up-regulated during and after cerebral ischaemia [11]. Zong et al [11] investigated the effects of RIPC on HIF1a, discovering that RIPC significantly reduced the expression of HIF1a when compared to the control group one day post reperfusion. These results correlated with a 10.3% reduction in cerebral infarct volume [11], suggesting that HIF1a is an important target in the neuro-protective effects of RIPC [11,12].
The mechanism in which HIF1a mediates its downstream effects is still a topic of some speculation [11,12]. Recent research by Koh et al [13] investigated how HIF1a interacts with another downstream signalling molecule, TIM3. It was found that in response to hypoxia, HIF1a binds to the TIM3 prompter region to increase its expression. Increased TIM3 up-regulates the inflammatory cytokines IL1b and CXCL1 (Figure 2) [13]. Blockage of TIM3 activity by monoclonal antibodies showed a significant reduction in the expression of both these cytokines as well as a substantial reduction in cerebral infarct volume. The findings of Zong et al [11] and Koh et al [13] suggest that RIPC exerts its neuro-protective effect via inhibition of HIF1a; subsequently resulting in down-regulation of TIM3 and reduction in cytokine and ROS production [10-13].
PKCd, JAK2/STAT 3 and P38 MAPK apoptotic pathways
Apoptosis is a process of organised cell death, triggered by an extensive number of signalling cascades. One well known example involves PKCd, an intracellular enzyme, activated by cell stress to initiate apoptosis [14]. It has been extensively reported that RIPC exerts an anti-apoptotic effect via reduction of PKCd expression [3,5,14]. However, recent research has investigated several additional pathways which may be involved in the anti-apoptotic effects of RIPC. One such pathway is the JAK2/STAT3 axis which has been found to attenuate apoptosis in multiple models of IRI [15,16]. Cheng et al [17] recently investigated the potential effects of RIPC on JAK2/STAT3, finding that it was associated with a significant increase in both JAK2 activity and STAT3 protein expression. The increase in STAT3 expression results in the transcription of anti-apoptotic Bcl2 family proteins, which inhibit the formation of Bax channels and the initiation of apoptosis [15-17].
As well as its effect on anti-apoptotic signalling, Cheng et al [17] also demonstrated that RIPC was potentially involved in the JAK2/STAT3 mediated inhibition of NFkB. The inhibition of NFkB results in the reduction of pro-inflammatory cytokines such as IL1b and TNFa; subsequently reducing recruitment of neutrophils and other leukocytes to the brain [3,17]. The collective effects of RIPC on JAK2/STAT3 mediated reduction of inflammation and apoptosis, resulted in a 13.2% reduction in rodent cerebral infarct volume when compared to the IRI control group (Figure 3) [17].

PI3k/Akt and eNOS uncoupling
Endothelial nitric oxide synthase (eNOS) is responsible for the physiological production of nitric oxide (NO), a key regulator of normal endothelial cell function. During IRI eNOS can become uncoupled and as a result transfer its electrons to an oxygen molecule to produce superoxide free radicals [22]. These superoxide radicals can cause direct cellular damage but can also combine with NO to produce peroxynitrite, a highly destructive reactive nitrogen species (RNS) which suppresses protein function in the same way as ROS [8,22].
The process of eNOS uncoupling is thought to result from both ROS mediated damage and the depletion of the eNOS substrate BH4 [22,23]. Chen et al [22] investigated the capacity of RIPC to attenuate these effects; finding that it was associated with both an increase in BH4 availability, as well as a reduction in ROS (through inhibiting transcription of NADPH oxidase and xanthine oxidase). The RIPC mediated inhibition of eNOS uncoupling resulted in a significant reduction in peroxynitrite levels, as well as a 15.1% reduction in cerebral infarct volume when compared to the control group [22].
RIPC has also been found to directly increase the synthesis of the eNOS enzyme, leading to higher levels of NO during reperfusion [22,24,25]. It is thought that this effect may be associated with the RIPC induced up-regulation of the PI3k enzyme, which triggers phosphorylation of the protein kinase B (Akt) transcription factor. Akt is an important anti-apoptotic signalling factor, which is also thought to upregulate eNOS transcription [24,25]. The increased production of eNOS increases the quantity of NO produced within the ischaemic brain; this promotes vasodilation and reduces inflammation and thrombosis [22,24]. Furthermore, the reduction in eNOS uncoupling increases the proportion of NO to superoxide radicals produced [22]. Hence, it appears that RIPC works synergistically to upregulate eNOS transcription whilst reducing uncoupling, resulting in an increase in NO and reduction in ROS and RNS [8,22-25].
Nrf-Antioxidant response element (ARE) pathway
Small amounts of ROS are produced in normal physiological processes, but cause no damage because they are rapidly neutralised by cellular antioxidants. The excessive ROS production in IRI overwhelms the antioxidant system and leads to extensive cellular damage [8,26]. Li et al [26] investigated the effects of RIPC on the Nrf2-ARE pathway. It was found that RIPC increased the activity of Nrf2 which subsequently increased ARE transcription [26]. ARE increases the production of the antioxidant molecules SOD, HO1, and NQO1 which, through a range of different cellular processes, neutralise oxygen free radicals into less reactive substrates (Figure 4) [27,28]. RIPC mediated neutralisation of ROS correlated with a significant reduction in MDA (a lipid peroxidation marker), as well as a 16.9% reduction in cerebral infarct volume compared to the control group [26].

AQP4 and ROS in formation of cerebral oedema
ATP deficiency during the period of ischaemic stroke triggers dysfunction of the Na+/K+ ATPase, accumulation of intracellular sodium, and influx of water into the astrocytes. Fluid accumulation within the astrocyte produces cytotoxic oedema, an event which results in death and destruction of nearby tissue [3,4]. Recent research has shown that this influx of water during IRI is likely mediated by the aquaporin 4 (AQP4) channels [29]. Li et al [30] has found that RIPC can reduce the expression of AQP4 channels in the brain following reperfusion, thus reducing the influx of water into the astrocytes. Reduction in astrocyte swelling was found to attenuate cytotoxic oedema formation, resulting in a 15.0% reduction in rodent cerebral infarct volume compared to the control group [30].
Cytotoxic oedema usually occurs in the earlier stages of IRI, when the blood-brain barrier (BBB) is still intact [3,4]. Later in disease progression, there is an increased production of ROS through the mechanisms which have previously been discussed [22,24,26,31]. ROS have been found to mediate BBB damage through oxidative damage, tight junction modification, and activation of matrix metalloproteinases (MMP) [32]. As a result of these events, BBB permeability increases, leading to influx of fluid into the neuronal extracellular space in an event called vasogenic oedema [31,32]. Li et al [30] demonstrated that RIPC may reduce BBB permeability and hence reduce vasogenic oedema: a finding supported by another study investigating RIPC in carotid stenosis [33]. RIPC most likely reduces BBB permeability through a variety of pathways targeting inflammation, ROS and antioxidant production that may all impact the integrity of the vascular endothelium or initiate endothelial dysfunction [29-33].
Integrating the pathways
RIPC is an expanding field with more pathways being discovered each year. As this occurs, it is important to know how each pathway relates to another (Figure 5).
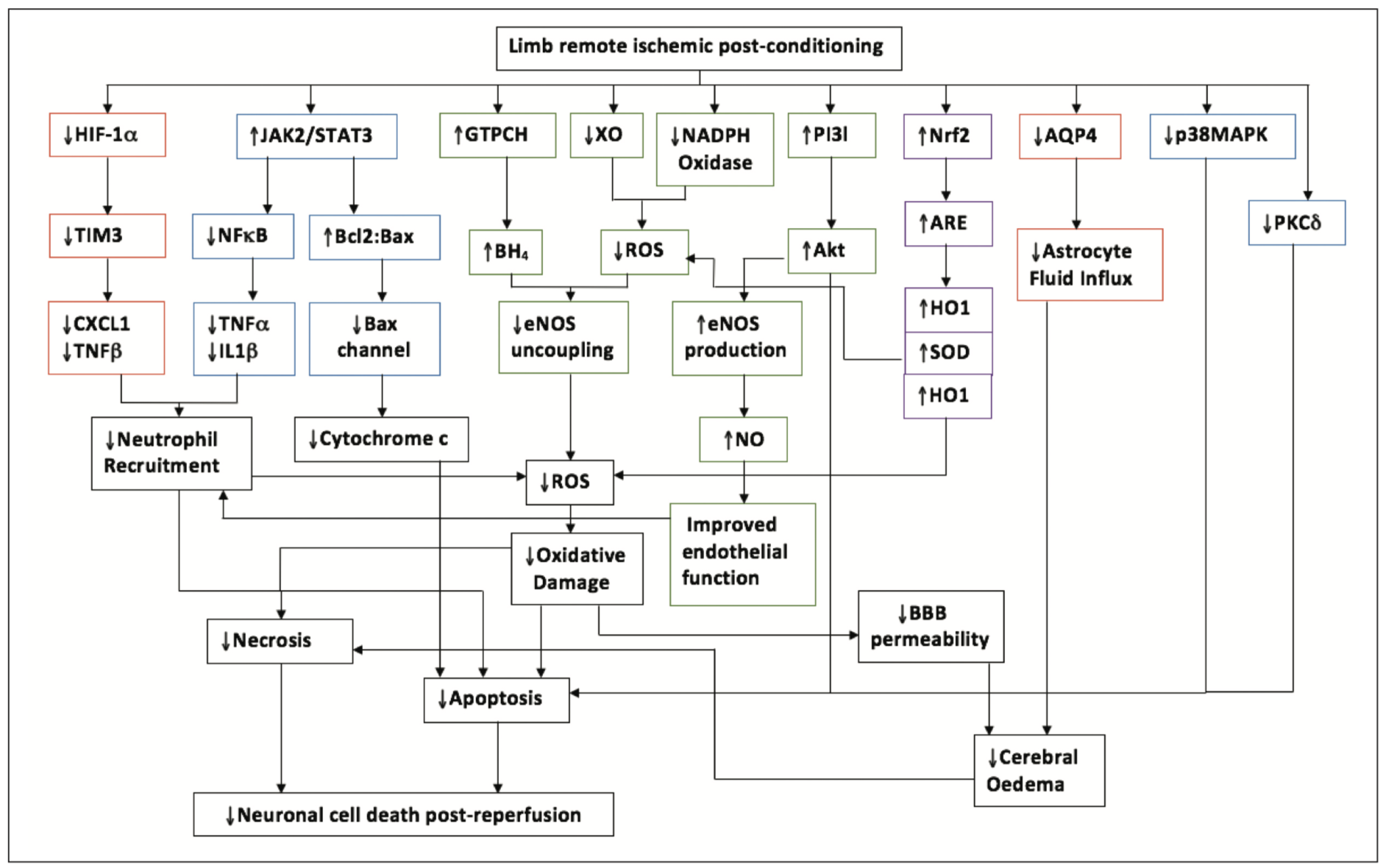
RIPC in the clinical setting
The application of RIPC in animal cerebral ischaemia models has been demonstrated to be safe and efficacious, reducing cerebral infarct volume and apoptotic neuronal populations substantially (Table 1) [5,7,34].
Whilst the preclinical evidence for RIPC is convincing, there is still a large gap in the translation to clinical trials. In May of 2017, the results of the RECAST trial were published. This phase I pilot blinded placebo controlled trial involved 26 patients with ischaemic stroke, 13 of whom were given the RIPC intervention within 24 hours of ischaemic stroke. Whilst small, this pilot study found that RIPC was very well tolerated, safe, and feasible in the clinical setting. Furthermore, RIPC was found to provide significant improvements in neurological function when compared to the control group [35]. As the only study published of its kind, the evidence for RIPC in stroke is still tentative, however, a recent meta-analysis on the use of RIPC in acute coronary syndrome has provided some more firm results. The analysis of 13 clinical trials demonstrated that RIPC was effective in reducing infarct size, reperfusion injury, and improving patient outcomes post myocardial infarction [36]. These recent publications provide a convincing argument for the further clinical exploration of how RIPC can be utilised in patients who suffer from ischaemic stroke.
Current gaps
Confusion surrounding the molecular mechanism of RIPC primarily pertain to a gap in understanding how an episode of ischaemia in the lower limb can result in an increased expression of neuro-protective factors within the brain [6]. Current research, predominantly conducted on cardiac models, suggests that this effect is likely achieved through multiple neurological, humoral, and immune events. How these mechanisms relate to ischaemic stroke and how they result in such diverse effects is still unknown [6,37-39]. Further research must be conducted, particularly in cerebral ischaemia models, before links can be made regarding the initiation of the RIPC neuroprotective pathways.
Conclusion
IRI is a major contributor to the death and destruction of the neuronal tissue after ischaemic stroke. The RIPC intervention aims to attenuate this damage through stimulation of neuroprotective pathways within the brain. Although clinical trials remain limited, RIPC has shown substantial efficacy in the pre-clinical setting: reducing cerebral infarct volume and apoptotic neuron populations. Research should continue to be conducted regarding the pathways involved in RIPC, however, a shift must also take place in translating this pre-clinical knowledge into clinical trials. The limited clinical data is positive thus far, but more must be done to determine whether this intervention is appropriate for the clinical setting.
Conflict of interest
None declared.
References
[1] Australian Institute of Health and Welfare. Stroke and its management in Australia: an update 2013 [Internet]. 2013 [cited 2016 Jun] Available from: http://www.aihw.gov.au/publication-detail/?id=60129543613.
[2] Nadel L. Stroke. Encyclopedia of cognitive science. Wiley; 2005.
[3] Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M. Molecular mechanisms of ischemia–reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 2013;47(1):9-23.
[4] Nour M, Scalzo F, Liebeskind DS. Ischemia-reperfusion injury in stroke. Interv Neurol. 2013;1(3-4):185-99.
[5] Wang Y, Reis C, Applegate nR, Stier G, Martin R, Zhang JH. Ischemic conditioning-induced endogenous brain protection: applications pre-, per- or post-stroke. Exp neurol. 2015;272:26.
[6] Ren C, Yan Z, Wei D, Gao X, Chen X, Zhao H. Limb remote ischemic postconditioning protects against focal ischemia in rats. Brain Res. 2009;1288:88-94.
[7] Hess DC, Hoda MN, Bhatia K. Remote limb perconditioning [corrected] and postconditioning: will it translate into a promising treatment for acute stroke? Stroke. 2013;44(4):1191-97.
[8] Dorweiler B, Pruefer D, Andrasi TB, Maksan SM, Schmiedt W, Neufang A, et al. Ischemia-reperfusion injury: pathophysiology and clinical implications. Eur J Trauma Emerg Surg. 2007;33(6):600-12.
[9] Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88(2):581-609.
[10] Boros P, Bromberg JS. New cellular and molecular immune pathways in ischemia/reperfusion injury. Am J Transplant. 2006;6(4):652-8.
[11] Zong Y, Jiang L, Zhang M, Zhou F, Qi W, Li S, et al. Limb remote ischemic postconditioning protects cerebral ischemia from injury associated with expression of HIF-1α in rats. BMC Neurosci. 2015;16:97-105.
[12] Shi H. Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Curr Med Chem. 2009;16(34):4593-608.
[13] Koh HS, Chang CY, Jeon S-B, Yoon HJ, Ahn Y-H, Kim H-S, et al. The HIF-1/glial TIM-3 axis controls inflammation-associated brain damage under hypoxia. Nat Commun. 2015;6:6340.
[14] Wang Q, Zhang X, Ding Q, Hu B, Xie Y, Li X, et al. Limb remote postconditioning alleviates cerebral reperfusion injury through reactive oxygen species-mediated inhibition of delta protein kinase C in rats. Anesth Analg. 2011;113(5):1180-7.
[15] Liu X, Zhang X, Zhang J, Kang N, Zhang N, Wang H, et al. Diosmin protects against cerebral ischemia/reperfusion injury through activating JAK2/STAT3 signal pathway in mice. Neuroscience. 2014;268:318-27.
[16] Zheng W-x, Wang F, Cao X-l, Pan H-y, Liu X-y, Hu X-m, et al. Baicalin protects PC-12 cells from oxidative stress induced by hydrogen peroxide via anti-apoptotic effects. Brain Inj. 2014;28(2):227-34.
[17] Cheng Z, Li L, Mo X, Zhang LU, Xie Y, Guo Q, et al. Non-invasive remote limb ischemic postconditioning protects rats against focal cerebral ischemia by upregulating STAT3 and reducing apoptosis. Int J Mol Med. 2014;34(4):957-66.
[18] Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15(1):11-8.
[19] Li H, Zhou S, Wu L, Liu K, Zhang Y, Ma G, et al. The role of p38MAPK signal pathway in the neuroprotective mechanism of limb postconditioning against rat cerebral ischemia/reperfusion injury. J Neurol Sci. 2015;357(1-2):270-5.
[20] Ma Q, Li X, Vale-Cruz D, Brown ML, Beier F, LuValle P. Activating transcription factor 2 controls Bcl-2 promoter activity in growth plate chondrocytes. J Cell Biochem. 2007;101(2):477-87.
[21] Liu W-H, Chang L-S. Arachidonic acid induces Fas and FasL upregulation in human leukemia U937 cells via Ca 2+/ROS-mediated suppression of ERK/c-Fos pathway and activation of p38 MAPK/ATF-2 pathway. Toxicol Lett. 2009;191(2):140-8.
[22] Chen G, Yang J, Lu G, Guo J, Dou Y. Limb remote ischemic post-conditioning reduces brain reperfusion injury by reversing eNOS uncoupling. Indian J Exp Biol. 2014;52(6):597.
[23] Gulati P, Singh N. Pharmacological evidence for connection of nitric oxide-mediated pathways in neuroprotective mechanism of ischemic postconditioning in mice. J Pharm Bioallied Sci. 2014;6(4):233-40.
[24] Peng B, Guo Q-l, He Z-j, Ye Z, Yuan Y-j, Wang N, et al. Remote ischemic postconditioning protects the brain from global cerebral ischemia/reperfusion injury by up-regulating endothelial nitric oxide synthase through the PI3K/Akt pathway. Brain Res. 2012;1445:92-102.
[25] Gulati P, Singh N. Evolving possible link between PI3K and NO pathways in neuroprotective mechanism of ischemic postconditioning in mice. Mol Cell Biochem. 2014;397(1):255-65.
[26] Li P, Su L, Li X, Di W, Zhang X, Zhang C, et al. Remote limb ischemic postconditioning protects mouse brain against cerebral ischemia/reperfusion injury via upregulating expression of Nrf2, HO-1, NQO-1 in mice. Int J Neurosci. 2015:1-28.
[27] Dinkova-Kostova AT, Talalay P. NAD(P)H: quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys. 2010;501(1):116-23.
[28] Shah ZA, Nada SE, Doré S. Heme oxygenase 1, beneficial role in permanent ischemic stroke and in Gingko biloba (EGb 761) neuroprotection. Neuroscience. 2011;180:248-55.
[29] Akdemir G, Ratelade J, Asavapanumas N, Verkman AS. Neuroprotective effect of aquaporin-4 deficiency in a mouse model of severe global cerebral ischemia produced by transient 4-vessel occlusion. Neurosci lett. 2014;574:70-5.
[30] Li S, Hu X, Zhang M, Zhou F, Lin N, Xia Q, et al. Remote ischemic post-conditioning improves neurological function by AQP4 down-regulation in astrocytes. Behav Brain Res. 2015;289:1-8.
[31] Pun PBL, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43(4):348-64.
[32] Freeman L, Keller J. Oxidative stress and cerebral endothelial cells: regulation of the blood-brain-barrier and antioxidant based interventions. Biochim Biophys Acta. 2012;1822(5):822-9.
[33] Yang F, Zhang X, Sun Y, Wang B, Zhou C, Luo Y, et al. Ischemic postconditioning decreases cerebral edema and brain blood barrier disruption caused by relief of carotid stenosis in a rat model of cerebral hypoperfusion. PLoS One. 2013;8(2):e57869.
[34] Ma XD, Song JN, Zhang M, An JY, Zhao YL, Zhang BF. Advances in research of the neuroprotective mechanisms of cerebral ischemic postconditioning. Int J Neurosci. 2015;125(3):161-9.
[35] England T, Hedstrom A, O’Sullivan S, Donnelly R, Barrett D, Sarmad S, et al. RECAST (Remote Ischemic Conditioning After Stroke Trial) a pilot randomized placebo controlled phase ii trial in acute ischemic stroke. Stroke. 2017;48:1412-5.
[36] Man C, Gong D, Zhou Y, Fan Y. Meta-analysis of remote ischemic conditioning in patients with acute myocardial infarction. Sci Rep. 2017;7:43529.
[37] Weber C. Far from the Heart: Receptor cross-talk in remote conditioning. Nat Med. 2010;16(7):760-2.
[38] Gho BC, Schoemaker RG, van den Doel MA, Duncker DJ, Verdouw PD. Myocardial protection by brief ischemia in noncardiac tissue. Circulation. 1996;94(9):2193-200.
[39] Kerendi F, Kin H, Halkos ME, Jiang R, Zatta AJ, Zhao ZQ, et al. Remote postconditioning: brief renal ischemia and reperfusion applied before coronary artery reperfusion reduces myocardial infarct size via endogenous activation of adenosine receptors. Basic Res Cardiol. 2005;100(5):404-12.